hemofilija
Sinonimi
Hemofilija, dedna motnja krvavitve, pomanjkanje faktorja strjevanja krvi,
Pomanjkanje faktorja VIII, pomanjkanje faktorja IX, hemofilija
opredelitev
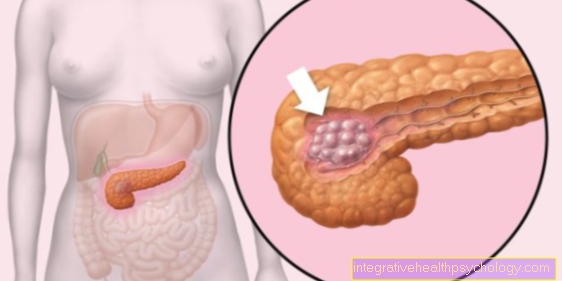
Hemofilija je dedna bolezen sistema strjevanja krvi:
Prizadeti bolniki imajo moteno strjevanje krvi, kar se kaže v dejstvu, da se krvavitev pojavi z najmanjšo travmo in krvavitev težko ustavi.
Koagulacijskih faktorjev ni mogoče aktivirati, tako da redni potek koagulacijske kaskade ni mogoč.
Hemofilija je prirojena bolezen, ki se pojavlja v dveh oblikah,
Lahko sta prisotni hemofilija A in B.
Hemofilija A se pojavlja pogosteje (v 85% primerov) kot oblika B (približno 15%) in je resnejša od obeh oblik.
Dejavnost kompleksa faktorja VIII je pri hemofiliji A omejena, ker se tvorba faktorja VIII-C v jetrnih celicah zmanjša.
Faktor VIII-C je ena od dveh podenot kompleksa faktorja VIII:Willebrand-Faktor in faktor VIII-C tvorita ta kompleks, ki pospešuje koagulacijo v koagulacijski kaskadi. Faktor von Willebrand ščiti faktor VIII-C pred hitro razgradnjo.
Količino funkcionalnega faktorja VIII se posledično zmanjša v obliki A.
V hemofilija B. sinteza koagulacijskega faktorja IX poteka v manjši meri, tako da je njegov učinek v procesu koagulacije odsoten in hemostaza oslabljena.
Pojav / epidemiologija
Pojav hemofilije v populaciji je za hemofilijo A. 1 od 5.000 in za obliko B. 1:15.000.
Dejstvo, da zlasti moški zbolijo, je posledica vprašanj, povezanih s spolom
X-vezana oblika dedovanja koagulacijske okvare (glej vzrok / razvoj).
Osnove
Koagulacija krvi je natančno nastavljen sistem: različne sestavine krvi, tako imenovani koagulacijski faktorji, se aktivirajo drug za drugim na kaskaden način, da se omogoči hemostaza v primeru poškodbe.
Koagulacijski sistem je sestavljen iz primarne in sekundarne hemostaze, ki ji pravimo tudi plazmatska koagulacija.
Če se poškoduje stena plovila, se prizadeta žila v prvem koraku zoži (= primarna koagulacija). Nato je podpora (=Trombocitna tromba) na poškodovanem območju, ki je prekrit s trombociti (= Trombociti) je oblikovan tako, da doseže začetno tesnjenje plovila.
Tvorba tromba aktivira plazemsko koagulacijo:
Začne se kaskada koagulacije in interakcija različnih dejavnikov koagulacije ustvari stabilno zapiranje rane ali ožilja.
Vzrok in izvor
The hemofilija Obe obliki hemofilije sta podedovani kot X-vezana recesivna lastnost.
V tako imenovanem X-povezanem načinu dedovanja hemofilije je genetska informacija za gensko zdravilo na spolnem kromosomu X, ki ga najdemo v dvojnih številkah pri ženskah in v enotnih številkah pri moških.
Za vsako gensko informacijo obstajata dva gena, tako imenovani aleli. Če pride do recesivnega dedovanja, morata biti na alele prizadeta mutacija, da se povzroči bolezen: recesivna pomeni, da patološko spremenjena genetska informacija izhaja iz zdravega, tj. se ne spremeni, informacije se zatirajo in ne vodijo v bolezen. Recesijska dedna bolezen se razvije le, če oba alela prenašata napačne genetske informacije.
Je samo a Izraz gena se je spremenil, pridite Ne do hemofilija.
Informacije: dedovanje
Če spremenimo samo en genski izraz, bolezni ne bo.
Ker imajo moški samo en kromosom X v paru spolnih kromosomov in so zato informacije o genskih izdelkih na njihovem X kromosomu na voljo le v preprosti obliki, bodo zboleli, če je pokvarjen le en alel.
Po drugi strani ženske, ki imajo dva kromosoma X kot spolni kromosom, zbolijo le, če imata oba kromosoma napačne gene za genski produkt.
Pri moških, ki trpijo za hemofilijo, je alel, ki nosi dedne podatke o faktorju koagulacije VIII-C (hemofilija A) ali koagulacijskemu faktorju IX (hemofilija B), na voljo le v nekoliko spremenjeni obliki;
Ženske morajo nositi to gensko spremembo na obeh alelih, da so bolne.
Če imajo ženske okvarjen in nespremenjen alel, jih imenujemo nosilke dedne bolezni, povezane s X, kot je hemofilija.
Pri hemofiliji imajo napačne genetske informacije učinek zmanjšanja aktivnosti prizadetega faktorja strjevanja ali zmanjšanja produkcije koagulacijskega faktorja.
X kromosom je nosilec genskega materiala za faktorje strjevanja zaradi sekundarne hemostaze.
Približno 70% mutacij v genskem materialu, ki povzročajo hemofilijo, se prenašajo v družinah, 30% bolezni izhaja iz spontanih sprememb (= spontanih mutacij) v genetskih informacijah.
diagnoza
Po tem, ko ste se pozanimali o bolnikovi anamnezi in opravili temeljit fizični pregled, so na voljo dodatni koraki za diagnozo hemofilije:
V 2/3 primerov gre za primere hemofilije v družini, zato je treba povprašati o družinskem združevanju bolezni, če bolnik zdravniku predstavi simptome, sumljive na hemofilijo.
Bolniki poročajo o modricah kot posledica najmanjših poškodb.
Eden jih razlikuje Resnost hemofilije v blago, zmerno ali hudo obliko bolezni.
Pregled krvnega vzorca zagotavlja naslednje rezultate, značilne za hemofilijo:
Čas krvavitve je normalen (= primarna koagulacija je nedotaknjena), vendar se funkcija plazemske koagulacije zmanjša, zato je tako imenovani čas PTT daljši.
PTT pomeni delni tromboplastinski čas. Merimo ga v laboratorijskih preskusih za preverjanje delovanja faktorjev I, II, V, VIII do XII ter XIV in XV.
Ker pri hemofiliji manjka en dejavnik koagulacijske kaskade, ne more teči na optimalen način, kar ima za posledico daljši čas koagulacije.
Da bi lahko razlikovali hemofilijo A in B, je treba bolnikovo kri pregledati zaradi faktorjev VIII in IX; odsotnost obeh dejavnikov določa vrsto hemofilije.
Diferencialna diagnoza
Hemofilijo je treba razlikovati od drugih motenj koagulacijskega sistema, pri čemer je zlasti omenjen sindrom von Willebrand-Jürgens.
Faktor von Willebrand (vWF) deluje skupaj s faktorjem VIII-C im
Kompleks faktorja VIII povzroči inhibicijo prezgodnje razgradnje faktorja VIII, poleg tega dejavnik spodbuja koagulacijo s posredovanjem adhezije krvnih ploščic na poškodovanem žilnem mestu. Tako je vWF pomemben del tako primarne kot sekundarne koagulacije.
Če je prisoten sindrom von Willebrand-Jürgens, potem nastanek
VWF v celicah stene žil poteka le v majhni meri ali na napačen način. Zmanjšana stopnja izobrazbe ali neustrezna funkcija študenta
von Willebrandov dejavnik povzročajo mutacije v genu za tvorbo faktorjev. Dedovanje sindroma ali mutacije vzročne gene se podeduje na avtosomno prevladujoč način: genetska informacija za vWF je na kromosomu številka 12, ki ni spolni kromosom. V prevladujočem načinu dedovanja oboleli alel zavira učinek drugega, zdravega alela, tako da bolezen povzroči mutacija gena in povzroči klinične simptome.
Bolniki običajno trpijo zaradi krvavitev kože in sluznice, manj zaradi spontanih krvavitev, kot pri bolnikih s hemofilijo.
Bolezen pogosto opazimo šele, ko je po operaciji dolgotrajna krvavitev.
Čas krvavitve pacienta po poškodbi se podaljša in vrednosti plazmatske koagulacije se patološko spremenijo (podaljšano PTT).
Sindrom von Willebrand-Jürgens povzroča dajanje desmopresina ali nadomestitev faktorja VIII in vWF, da se stabilizira koagulacija (podrobnejša razlaga glej pod "Terapija hemofilije").
Kakšni so simptomi hemofilije?
Simptomi v oblikah hemofilije se ne razlikujejo:
- Hemofilija običajno vodi le do kliničnih simptomov pri moških.
- Prekomerna krvavitev se pojavi s hemofilijo, kar ni v povezavi s prejšnjo, večinoma banalno nesrečo (travma), čas krvavitve je normalen, vendar se običajno pojavi krvavitev, ki se pri zdravih ljudeh ne pojavi.
- Razlikujemo lahko med tremi oblikami hemofilije, ki jih opredeljuje preostala aktivnost prizadetih koagulacijskih faktorjev:
1.huda hemofilija (hemofilija) z manj kot 1% preostale aktivnosti ali dela funkcionalnega dejavnika, ki je še vedno prisoten, pri katerem pride do spontane krvavitve in sklepne krvavitve
2. Zmerna hemofilija (krvna bolezen) s faktorsko aktivnostjo med
1 in 5% normalne faktorske aktivnosti in pojav modric (= hematomi) po manjši travmi
3. blaga hemofilija (hemofilija), pri katerih je še vedno 5–15% preostale aktivnosti in pri katerih bolniki poročajo o simptomih, kot so hematomi (modrice) po pomembni travmi in pooperativni krvavitvi.
Preberite več o temi: Kateri so vzroki za možgansko krvavitev
- Ženske, ki imajo običajno spremenjene genetske podatke za hemofilijo, običajno ne kažejo kliničnih simptomov z več kot 50% preostale aktivnosti.
- Bolniki doživljajo krvavitev v velike sklepe koleno-, Kolk- ali Rameni sklep, pri čemer eden govori o hemarthrosis. Krvavitev sproži vnetno reakcijo s popravljalnimi procesi v sklepu, kar lahko vodi v otrdelost sklepa.
- Lahko krvavi tudi v Muskulatura in mehka tkiva prihajajo. Krvavitev vodi v zvišanje tlaka v mišicah ali tkivu, saj je zdaj več volumna. To lahko vpliva zlasti na roke in noge Komoralni sindrom voditi:
Povišan tlak povzroči stiskanje krvnih žil in živcev, tako da so okončine premajhne in velike površine tkiva lahko umrejo. Prekomerni sindrom mora kirurg zdraviti čim prej, da prepreči izgubo okončine. - Pojavi se krvavitev v trebuh, kar je za bolnika smrtno nevarna situacija.
- Po operaciji je možna nenavadno dolgotrajna krvavitev.
Poleg tega lahko pride do daljših obdobij s krvjo v urinu. Tukaj eden grozi Anemijaker bolnik nenehno izgublja kri, morda neopaženo. - So še posebej nevarne Cerebralna krvavitev (= intrakranialna krvavitev), kar pri 10% ljudi s hemofilijo povzroči smrt. Več o tej temi najdete v naši temi Cerebralna krvavitev.
Kako poteka zdravljenje hemofilije?

Pri bolnih naj bi se pojavile krvavitve hemofilija Izogibati se je treba, zato pacient ne Zdravila ki zavirajo strjevanje krvi, kot so Acetilsalicilna kislina (Aspirin®) in brez intramuskularnih injekcij (= brizge v Muskulatura) oz.
Če pride do travme s krvavitvami, je skrbna lokalna hemostaza zelo pomembna, da preprečimo krvavitev v sosednje tkivo.
Zdravljenje z zdravili hemofilija sestoji iz nadomestitve koagulacijskih faktorjev, ki jih telo ne more proizvesti v zadostnih količinah.
Bolniki z rahlo hemofilija po potrebi prejemajte pripravke faktorja strjevanja, tj. če je prišlo do travme s krvavitvami ali če je načrtovana večja operacija, kar lahko povzroči obilno krvavitev.
Nadomestilo profilaktičnega faktorja je treba uporabiti pri bolnikih z zmerno do hudo hemofilijo, tj. manjkajoči dejavnik je treba dati stalno, prej pojavi se krvavitev.
Če je preostala aktivnost faktorja VIII večja od 15% ali faktorja IX več kot 20-25%, redno zdravljenje ni potrebno; ti bolniki prejmejo manjkajoči koagulacijski faktor v primeru spontane krvavitve ali pred načrtovanim operativnim posegom.
Dolgotrajna terapija kot tudi tista pred operacijo lahko poteka v obliki samozdravljenja doma, pri katerem bolnik sam uporabi manjkajoči koagulacijski faktor.
Za bolnike z blago hemofilijo obstaja alternativa za zdravljenje:
Zdravilna učinkovina Desmopresin (npr. Minirin®) vodi do porazdelitve
Faktor VIII, ki je shranjen v stenah posode.
Vendar pa lahko zdravilo dajemo le nekaj dni hkrati, saj se po stimulaciji sproščanja faktorja spomin v stenah žil izčrpa in ga je treba znova ponoviti.
V akutni terapiji hemofilija obstajajo tri možnosti intervencije:
- Bolnik prejme aktivirani protrombin, snov, ki pospešuje strjevanje krvi, tako da se kri čim hitreje ustavi.
- Druga terapevtska možnost je uporaba pripravkov faktorja VII. Ta dejavnik je na začetku kaskade koagulacije in začne svoj redni potek.
- Tretja terapevtska možnost je uporaba živalskega faktorja VIII-C, ki omogoča pacientu zaustavitev krvavitve.
Zapleti
Nadomestno darilo (=zamenjava) koagulacijskih faktorjev lahko privede do tvorbe protiteles proti ravno tem dejavnikom, tako da substitucija v stalnem odmerku nima več terapevtskih učinkov, zato govorimo o tvorbi zaviralcev.
Glede na določitev koncentracije protiteles v pacientovi krvi lahko dajemo visoke odmerke faktorja VIII, katerih cilj je ponovno pridobiti toleranco na ta dejavnik in izklopiti tvorbo protiteles. Ta postopek je treba izvesti le v specializiranih centrih.
Obstaja majhno tveganje za okužbo s hepatitisom ali okužbo z virusom HIV (=HIV), saj dejavniki izhajajo iz človeških krvnih pripravkov.
Drugo tveganje nadomestnega zdravljenja hemofilije je pojav alergijskih reakcij.